RNase H-dependent PCR (rhPCR) primers
Precise amplification of target sequences in applications such as rare variant identification, splice variant discovery, and microbial identification
rhPCR is a novel nucleic acid amplification technology that provides improved accuracy over traditional PCR. rhPCR primers (rhPrimers), unique primers that contain RNA bases, are used in conjunction with the thermostable RNase H2 enzyme to perform rhPCR.
Ordering
- Reduce the formation of primer-dimers or misprimed PCR products
- Reliably multiplex a large number of DNA targets
- Select from alternate rhPrimer designs to best suit desired application
In addition to your choice of Taq DNA polymerase and other general PCR reagents, rhPCR technology requires the following components:
- rhPCR Primers—use instead of conventional PCR primers
- RNase H2 enzyme—needed for activation of rhPCR Primers and PCR product extension
rhPCR Primers
Designed specifically for use with RNase H2. Primers include an RNA base and a blocking moiety at the 3′ end. Prices shown are for primers up to 60 bases with standard desalting.
Key: D = DNA base, match to target; r = RNA base, match to target; M = DNA base, mismatch to target; x = C3 space
* Standard desalting is sufficient for most situations and is included in the price of each primer. For experiments with long primers or requirements for above average purity, RNase-free HPLC purification is available for an additional fee.
Product Details
rhPCR requires uniquely designed primers (rhPrimers) and a thermostable RNase H2 enzyme. We offer two versions of rhPrimers. rhPrimer GEN1 designs may be used for genotyping and multiplexed amplification. For more demanding studies, such as rare variant identification, rhPrimer GEN2 designs are recommended.
rhPrimer design considerations
Efficient cleavage of a blocked primer by RNase H2 requires a footprint of at least 8–10 bases upstream of the single RNA base in the primer and 4 bases downstream of the RNA base. This footprint should be perfectly complementary to the template
intended for amplification. Mismatches can significantly reduce the efficiency of cleavage, especially when close to the RNA cleavage site.
A blocking group (represented by x in our design nomenclature) is used either to directly
block extension or to prevent replication in subsequent cycles. Typically, a C3 spacer is used as the blocking moiety in rhPCR primers.
Two versions of rhPrimers, GEN1 and GEN2, have been developed. These have different properties
and indications:
- The first-generation primer (rhPrimer GEN1) is represented by DDDDDDDDrDDDDMx, where D represents a DNA base, r represents the RNA base, M represents a mismatched DNA base, and x represents the blocker (usually a C3 Spacer). GEN1 primers are most appropriate for standard genotyping applications and for multiplexed amplification. This primer design works well with low levels of RNase H2 enzyme.
- The second-generation primer (rhPrimer GEN2) is represented by DDDDDDDDrDxxDM, where D represents a DNA base, r represents the RNA base, x represents the blocker, and M represents a mismatched DNA base. GEN2 primers are most appropriate for rare-allele
identification or for applications where extremely high fidelity of template amplification is desired. GEN2 primers may require use of higher amounts of RNase H2 enzyme (range is 1–100X that needed for GEN1 primers; titration and optimization
need to be performed for each GEN2 primer set; for this reason, we recommend use of GEN1 primers for most needs).
In general, we recommend avoiding rU as the RNA base at the cleavage site of GEN2 primers. In GEN2 format, primers containing rU require more RNase H2 enzyme for efficient cleavage than primers containing rC, rG, or rA. If rU cannot be avoided because of genotyping or target sequence constraints, carefully titrate the concentration of the RNase H2 enzyme to achieve efficient cleavage of the rU primer while minimizing an excess of enzyme present for other primers in the reaction; the presence of excess enzyme will decrease the precision of cleavage.
Primer design instructions
Like any other amplification reaction, good rhPCR requires use of high-quality, properly designed primers.
- Select a “mature primer”, which is usually the same primer that you would normally use for standard PCR against the same target.*
- Add the following for the rhPCR primer type:
- rhPrimer GEN1—Add an RNA base followed by 4 matching DNA bases, 1 mismatched DNA base, and the C3 blocking group to the 3′ end of the primer (Figure 1).†
- rhPrimer GEN2—Add an RNA base followed by a DNA base, 2 C3 blocking groups, another DNA base, and 1 mismatched DNA base to the 3′ end of the primer (Figure 2).†
- Design the final mature primer to the same specifications you would normally use for standard PCR, ensuring that the Tm of the primer is correct for the reaction conditions. An anneal/extend reaction temperature of 60°C is often used; however, rhPCR is effective at a temperature range of 50–70°C.‡
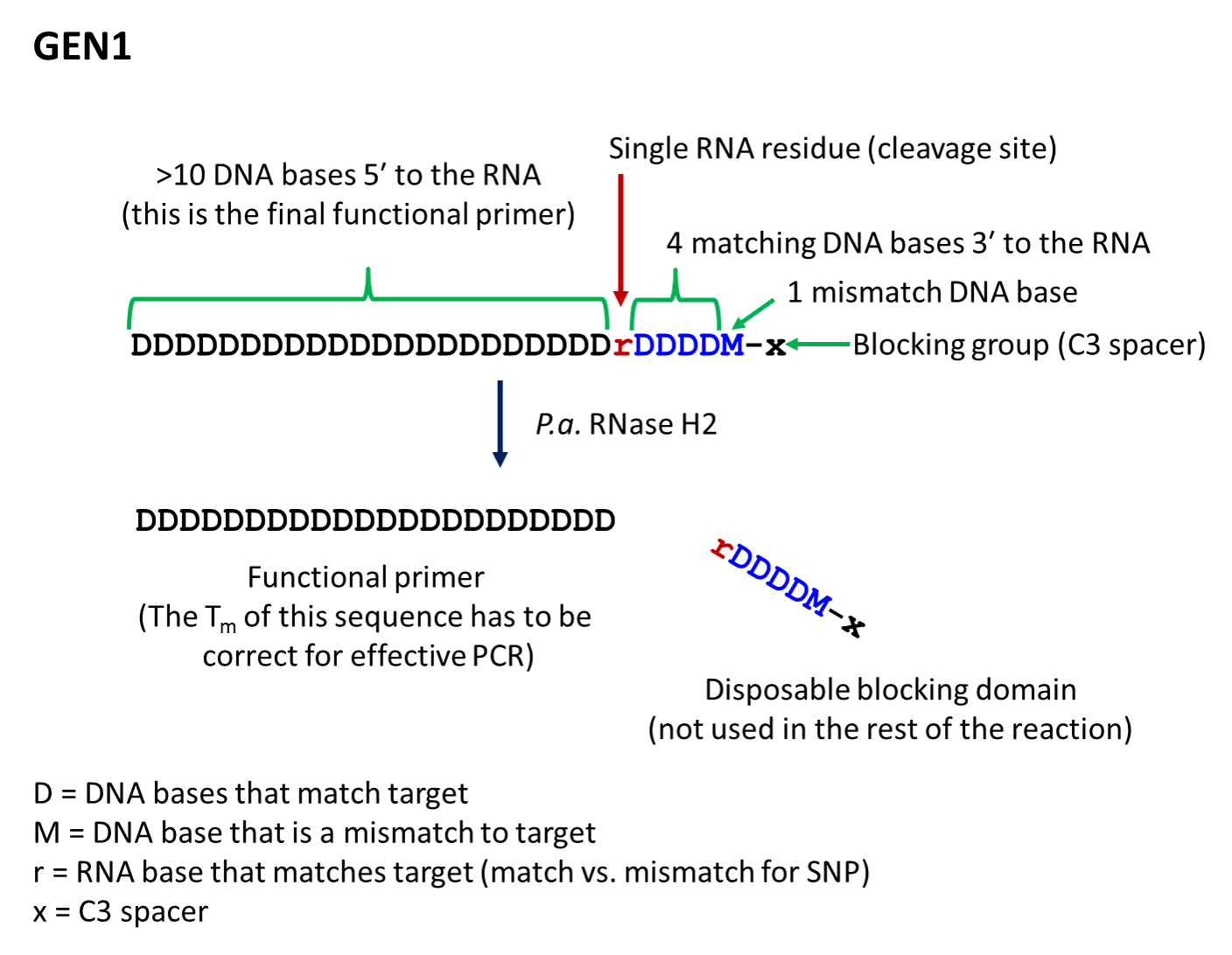
Figure 1. Design of rhPrimer GEN1.
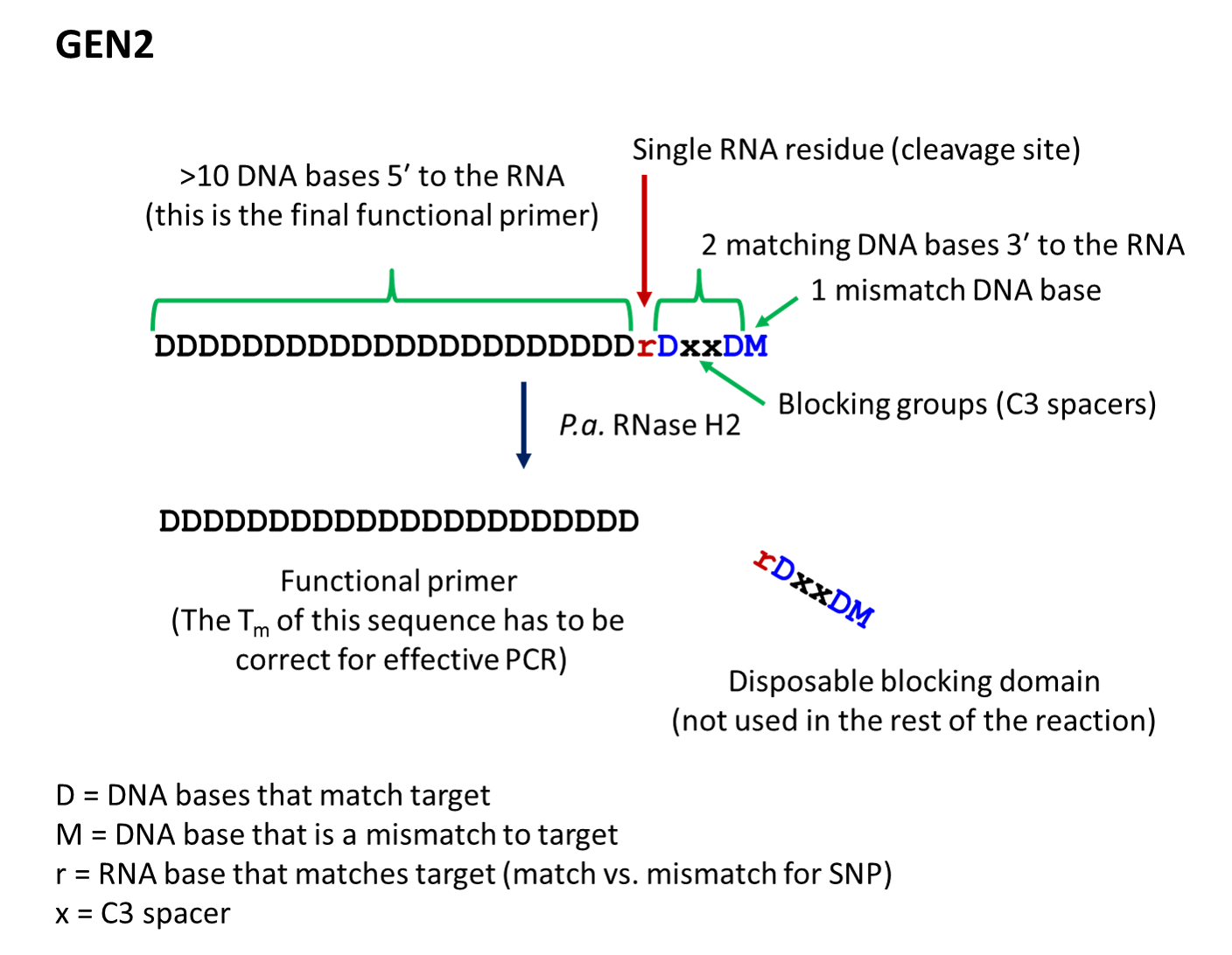
Figure 2. Design of rhPrimer GEN2.
* PCR primer pairs can be selected using the PrimerQuest™ Tool. Select qPCR 2 Primers Intercalating Dyes under Choose Your Design to get correct buffer conditions.
† The bases in the “disposable blocking domain” should be perfectly complementary to the target unless otherwise indicated ("M"). Mismatches placed closer to the RNA base will decrease the efficiency of cleavage by RNase H2. If you are designing primers for SNP identification, position the SNP at the RNA base, where the mismatch will have the greatest impact on RNase H2 cleavage.
The primer domain can be longer than shown. For example, a target-specific 3′ domain can be combined with a 5′-domain that is used as a universal primer binding site to permit universal amplification after RNase H2 cleavage or subsequent capture by universal capture probes.
‡ Primer Tm values can be calculated using the OligoAnalyzer™ Tool. It is important to input the buffer composition into the design tool so that correct Tm values are calculated. If you are using a commercial PCR master mix and do not know the buffer composition, use the values: 50 mM KCl, 3 mM MgCl2, and 0.8 mM dNTPs, which will approximate the conditions used in most qPCR master mix recipes.
How rhPCR works
High precision is often achieved with PCR; however, sometimes it is necessary to position primers at suboptimal locations in the target. This can result in the formation of primer-dimers and/or undesired amplification of homologous sequences.
IDT scientists have developed RNase H2–dependent PCR, a method for increasing PCR precision and reducing primer-dimers by using RNase H2 from Pyrococcus abyssi (P. abyssi) and DNA primers that contain a single ribonucleotide residue and a 3′ blocking moiety. The blocked primers are activated when cleaved by the RNase H2 enzyme. Cleavage occurs on the 5′ side of the RNA base after primer hybridization to the target DNA. Because the primers can only be cleaved after they hybridize to the perfectly matched target sequence, primer-dimers are reduced. The requirement for high target complementarity reduces amplification of closely related sequences (Figure 3).
Pyrococcus abyssi is an extreme thermophile, so the P. abyssi RNase H2 enzyme has optimal activity in range of 70–75°C and is functional in rhPCR between 50°C and 75°C. P. abyssi RNase H2 has very low activity at room temperature (~1000X less activity). Therefore, use of this enzyme to perform primer activation confers a "hot start" character to the PCR. P. abyssi RNase H2 is functional in most PCR buffers and can be added directly to the PCR master mix. The enzyme functions in real time, making the method a transparent change to standard PCR with primer cleavage occurring in the background during each anneal/extend cycle [1].
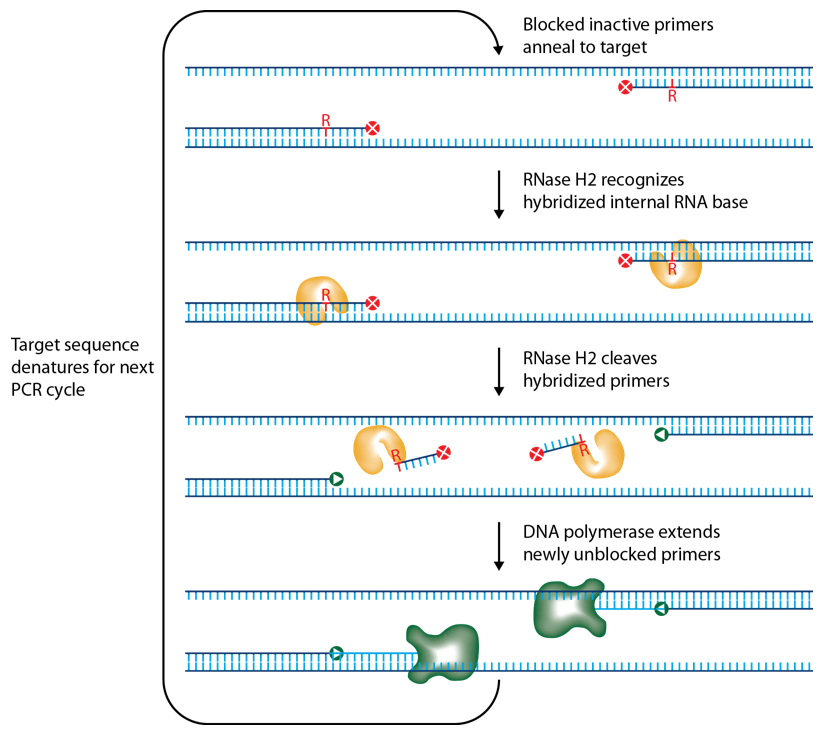
Figure 3. Schematic representation of rhPCR.
Thermostability
Pyrococcus abyssi RNase H2 can be incubated at 95°C for >45 minutes with little loss of activity (Figure 4) and, therefore, can survive PCR conditions.
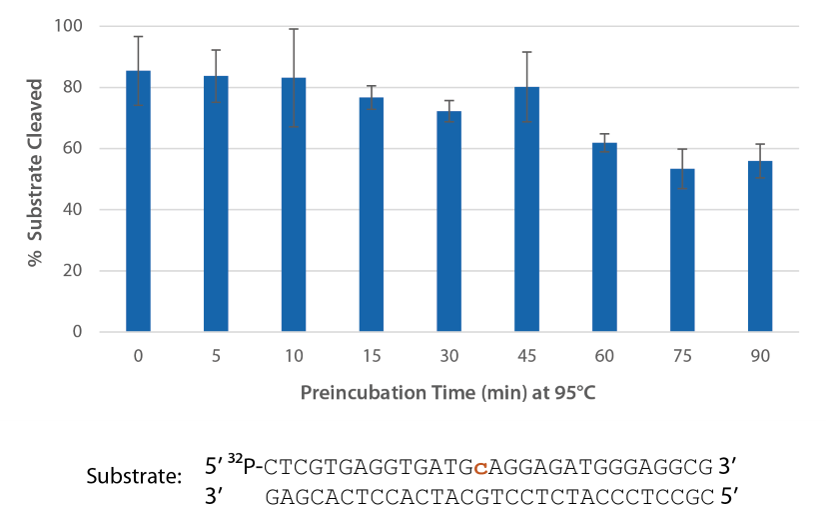
Figure 4. Thermostability of P. abyssi RNase H2 allows this enzyme to survive PCR conditions. P. abyssi RNase H2 was preincubated at 95°C for 0–90 min in buffer (10 mM Tris-HCl, pH 8.0; 50 mM NaCl; 0.01% Triton X-100; 10 µg/mL BSA; and 4 mM MgCl2). Radiolabeled substrate (2 pmol) was added to heat-treated RNase H2 (100 µU), and enzyme activity was assessed by measuring substrate cleavage after incubation at 70°C for 20 min. Error bars represent standard deviation (n = 3).
Compatibility with PCR temperatures
Pyrococcus abyssi RNase H2 is active at temperatures that are compatible with PCR conditions and inactive at room temperature (Figure 5).
- P. abyssi RNase H2 can be used to create “hot start” PCR conditions without the need for a modified “hot start” polymerase, because RNase H2 is essentially inactive at room temperature.
- You can optimize reaction conditions (e.g., increase the amount of RNase H2 per reaction) if your PCR cycling conditions require annealing/extension temperatures between 50–60°C (data not shown).
- P. abyssi RNase H2 activity reaches a plateau between 70–95°C (data not shown).
- Compared to other RNases, P. abyssi RNase H2 is relatively safe to have in laboratories that work with RNA, because this RNase H2 requires elevated temperatures, Mg2+, detergents, and duplexed RNA/DNA substrate for activity.
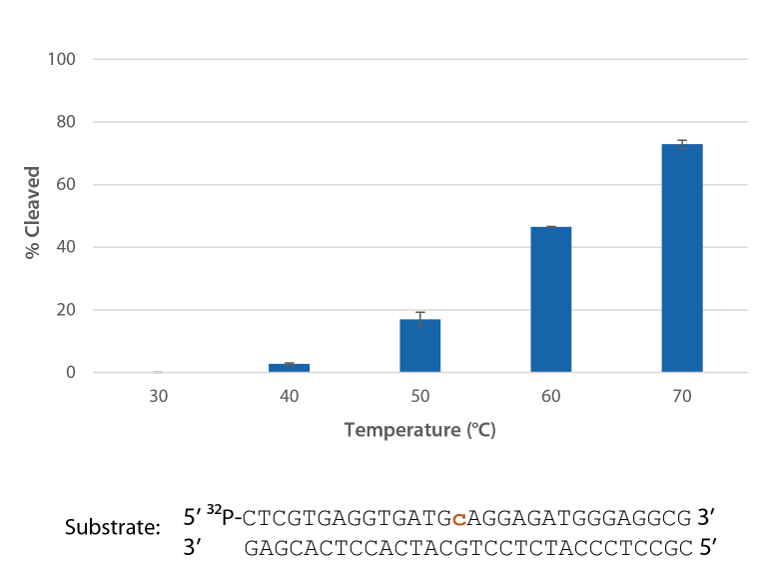
Figure 5. P. abyssi RNase H2 is active at temperatures compatible with PCR and inactive at room temperature (20–25°C). Enzyme activity of P. abyssi RNase H2 (250 µU) was assessed by measuring cleavage of a radiolabeled substrate (2 pmol) after incubation at varying temperatures for 20 min in buffer (10 mM Tris-HCl, pH 8.0; 50 mM NaCl; 0.01% Triton X-100; 10 µg/mL BSA; and 4 mM MgCl2). Error bars represent standard deviation (n = 3).
Compatibility with various Mg2+ concentrations used in PCR
Pyrococcus abyssi RNase H2 is active across a broad range of Mg2+ concentrations (Figure 6) that are compatible with PCR conditions (typically 3 mM).
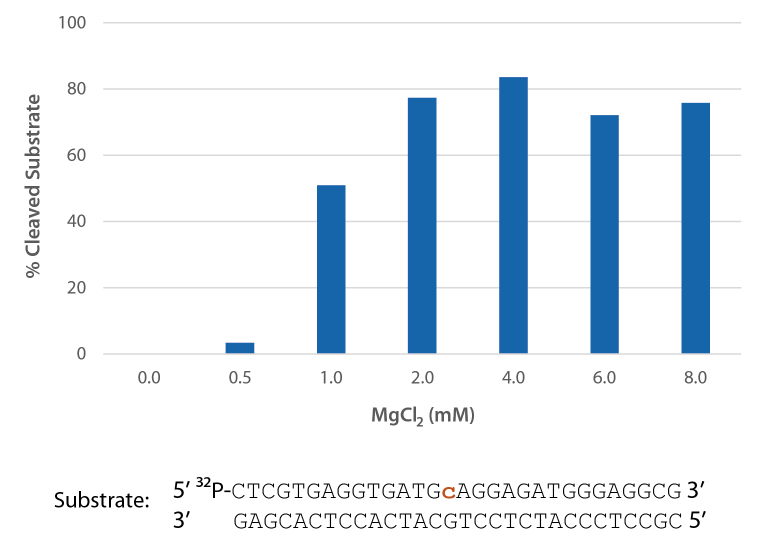
Figure 6. P. abyssi is active at Mg2+ concentrations compatible with PCR. Enzyme activity of P. abyssi RNase H2 (250 µU) was assessed by measuring cleavage of a radiolabeled substrate (2 pmol) after incubation at 70°C for 20 min in buffer (10 mM Tris-HCl, pH 8.0; 50 mM NaCl; 0.01% Triton X-100; and 10 µg/mL BSA), containing 0–8 mM MgCl2 (n = 2, per condition).
Detergent is required for optimal enzyme activity
The highest enzyme activity is observed with Triton™ X-100 and Tween® 20 (Figure 7).
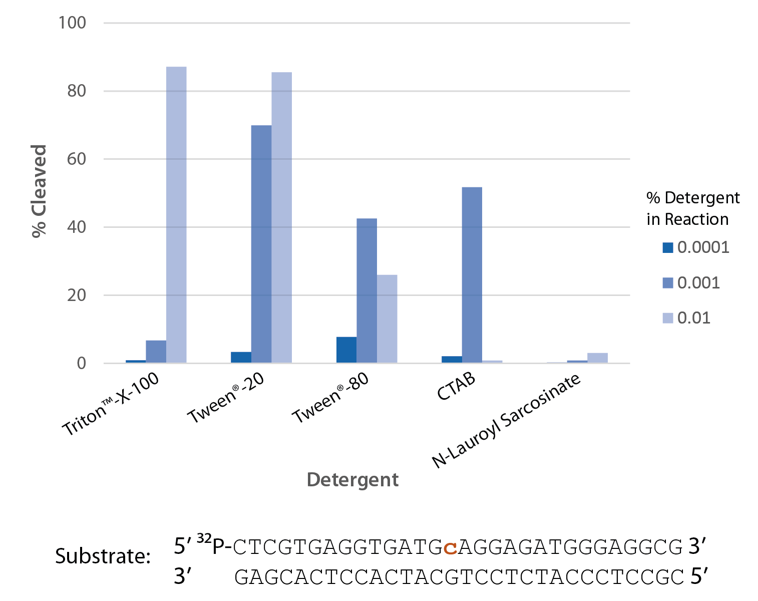
Figure 7. P. abyssi RNase H2 activity requires detergent. Enzyme activity of P. abyssi RNase H2 (100 µU) was assessed by measuring cleavage of a radiolabeled substrate (2 pmol) after incubation at 70°C for 30 min in buffer (10 mM Tris-HCl, pH 8.0; 50 mM NaCl; 10 µg/ml BSA; and 4 mM MgCl2), containing varying amounts of different detergents (n = 1 per condition).
Product Data
rhPCR significantly reduces primer-dimer artifacts
rhPCR uses activation of RNA-DNA hybrid primers by the RNase H2 enzyme from Pyrococcus abyssi. The primers are only activated upon precise annealing to the correct target, and this RNase H2 activation mechanism significantly reduces primer-dimers and other amplification artifacts that can affect experimental results. Figure 8 shows how effectively RNase H2 activation reduces primer-dimer artifacts, even when 96 primer pairs are multiplexed in a single PCR.
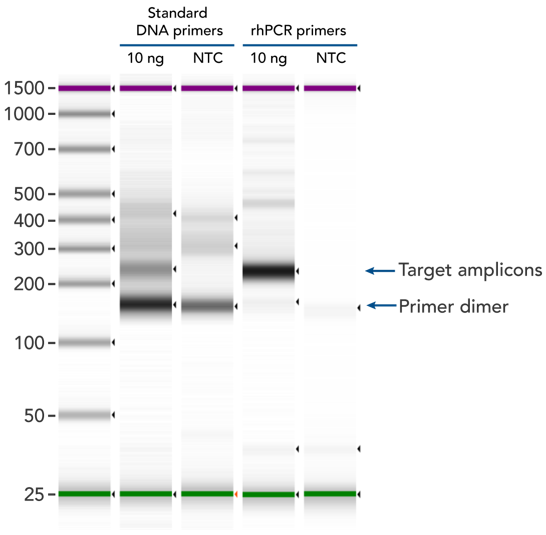
Figure 8. rhPCR primers significantly reduce primer-dimers and nonspecific amplification artifacts in multiplex applications. In multiplex PCR amplification of 96 targets from human genomic DNA (NA12878, Coriell Institute), two sets of multiplex primers for the 96 assays (192 individual primers) were synthesized either as standard PCR primers or as RNase H2–activated, rhPCR primer pairs. The results show tight control of primer-dimer artifacts, even in highly multiplexed assays. Total reaction volumes were 50 µL, with 10 ng of genomic DNA template or no template as controls. RNase H2 was present in all reactions, but has no functional role in PCRs with standard primers.